熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
TLR4激活被發現參與高葡萄糖誘導的系膜細胞過度增殖
蛋白質印跡顯示,高葡萄糖(25 mM)孵育顯著激活了TLR4的表達(圖1D)。通過siRNA下調TLR4可以逆轉小鼠系膜細胞的過度增殖(圖2C)。有趣的是,敲低CSE也顯著增加了小鼠系膜細胞中的TLR4表達(圖2B);NaHS處理可以顯著抑制TLR4激活并抑制細胞過度增殖(圖2B)。此外,TLR4下調并未顯著改變高葡萄糖誘導的CSE表達抑制(圖2A)。然而,TLR4沉默可以顯著消除由CSE下調引起的細胞過度增殖。這些結果暗示在由高血糖誘導的系膜細胞過度增殖通路中,TLR4可能位于CSE通路的下游。
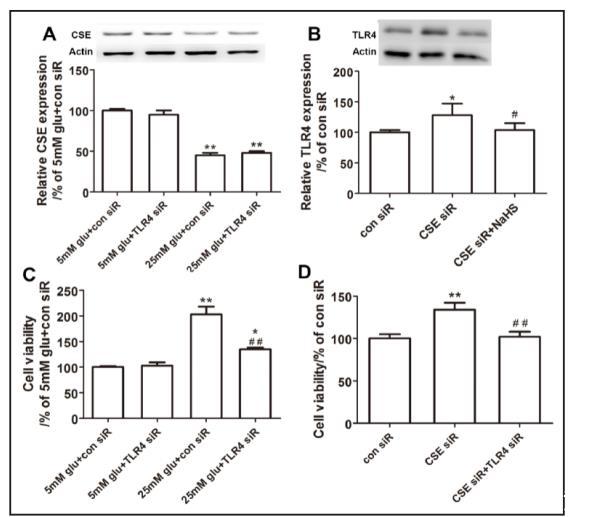
圖2. CSE抑制誘導的TLR4激活可能參與HG導致的系膜細胞過度增殖
(A) TLR4抑制未改變高葡萄糖處理的小鼠系膜細胞中CSE的表達(數值為均值±標準誤;P < 0.01 vs. 5 mM葡萄糖+對照siRNA;n = 4)。
(B) CSE抑制導致TLR4激活,而NaHS處理可逆轉這一現象(數值為均值±標準誤;*P < 0.05 vs. 對照siRNA;#P < 0.05 vs. CSE siRNA;n = 4)。
(C) TLR4沉默顯著逆轉HG誘導的細胞過度增殖(數值為均值±標準誤;P < 0.05,*P < 0.01 vs. 5 mM葡萄糖+對照siRNA;##P < 0.01 vs. 25 mM葡萄糖+對照siRNA;n = 4)。
(D) TLR4抑制顯著抑制CSE沉默誘導的系膜細胞過度增殖(數值為均值±標準誤;P < 0.01 vs. 5 mM葡萄糖+對照siRNA,##P < 0.01 vs. 25 mM葡萄糖+對照siRNA;n = 4)。
我們接著研究了TLR4對系膜細胞中PI3K-Akt通路的影響。蛋白質印跡分析顯示,高血糖顯著增強了PI3K和Akt的磷酸化,在TLR4敲低后恢復到正常水平,盡管PI3K和Akt的總表達保持不變(圖3A和B)。如圖3C所示,通過LY294002抑制PI3K也顯著減輕了高葡萄糖誘導的細胞增殖。
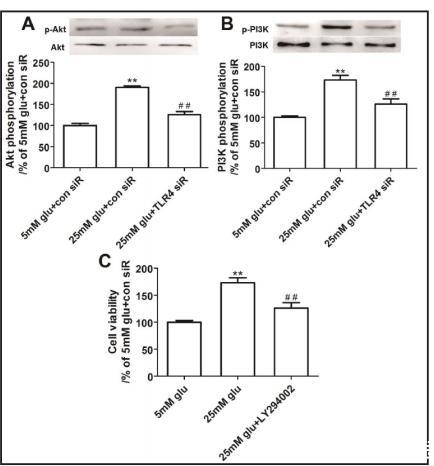
圖3. PI3K通路抑制以TLR4依賴性方式顯著抑制HG誘導的系膜細胞增殖
(A, B) HG導致小鼠系膜細胞PI3K/Akt表達顯著增加,而TLR4沉默可下調這一表達(數值為均值±標準誤;P < 0.01 vs. 5 mM葡萄糖+對照siRNA,##P < 0.01 vs. 25 mM葡萄糖+對照siRNA;n = 4)。
(C) PI3K抑制顯著抑制HG處理誘導的系膜細胞過度增殖(數值為均值±標準誤;P < 0.01 vs. 5 mM葡萄糖;##P < 0.01 vs. 25 mM葡萄糖;n = 4)。
下調TLR4抑制PI3K-Akt通路并抑制小鼠系膜細胞過度增殖
討論
本文的主要發現是,糖尿病腎損傷可能是由硫化氫的下調誘導的,這種下調依賴于TLR4表達增強和H?S合成減少。此外,TLR4介導的PI3K-Akt通路激活部分促進了糖尿病腎病的病理生理。
幾個世紀以來,硫化氫(H?S)一直被認為具有高度毒性。然而,最近H?S已被認為是介導廣泛生理效應的物質。一氧化氮(NO)是一種具有生物學和調節特性且半衰期很短(幾秒)的氣體,其特性與H?S相似。與NO一樣,H?S的合成和生物利用度對生理和病理生理過程至關重要。H?S是一種高親脂性分子,能夠通過擴散自由穿透所有類型細胞的膜,不需要專門的膜轉運蛋白。通過其存在和酶促產生,H?S作為重要的氣體遞質,如一氧化氮(NO)和一氧化碳(CO),因其信號傳導能力而受到關注。
越來越多的證據表明,H?S的上調對糖尿病引起的損傷(如心臟纖維化、腎小球足細胞損傷、傷口愈合障礙等)發揮保護作用。此外,硫化氫(H?S)的減少被認為在糖尿病血管并發癥(包括糖尿病腎病)中起關鍵作用。研究表明,較低的H?S濃度是糖尿病腎病發生和發展的原因。在伴有腎病的2型糖尿病中,H?S生成與CSE/CBS mRNA表達呈負相關。糖尿病相關腎病中存在紊亂的H?S代謝。此外,據報道H?S對認知疾病具有神經保護作用。H?S保護作用的潛在機制涉及氧化應激和自噬。
糖尿病腎病是世界范圍內終末期腎病的主要原因。顯著的系膜基質擴張是糖尿病患者腎臟病變的特征。不僅是系膜細胞,上調的硫化氫也能減輕高葡萄糖誘導的小鼠足細胞(MPC)損傷。然而,H?S發揮其多種作用的確切機制和通路尚未完全闡明。我們的研究結果表明,通過添加外源性供體(NaHS)或過表達CSE/CBS,上調H?S可以抑制小鼠系膜細胞的過度增殖。
TLR是病原體相關分子模式(PAMPs)受體家族之一,是具有胞質尾部的完整膜糖蛋白,在病原微生物的早期識別和宿主防御中發揮作用。TLR-4也識別其他配體。與其他TLR一樣,TLR-4能夠識別內毒素(LPS)和微生物分子(糖脂、呼吸道合胞病毒融合蛋白和熱休克蛋白)以及內源性損傷誘導的組織分子,從而產生細胞內信號響應。值得注意的是,高血糖被證明可誘導TLR4表達。Pahwa等人發現TLR4介導高血糖誘導的大血管主動脈內皮細胞炎癥和內皮糖萼的擾動。高血糖也能在糖尿病視網膜病變的內皮細胞中誘導并激活TLR4。本研究證明TLR4敲低抑制了由高血糖誘導的系膜細胞異常增殖,這與其他人的報道一致。
有趣的是,TLR4被報道與H?S合成和生物利用度相關。Tamizhselvi等人報道H?S可能通過P物質上調TLR4通路。與此一致,我們的體外實驗表明,下調H?S能顯著激活小鼠系膜細胞中的TLR4表達。反之,據報道在大鼠中TLR4通過NF-κB激活上調CBS表達。Zheng等人發現,與野生型小鼠相比,TLR4基因敲除小鼠顯示出H?S合成酶CSE的表達升高,從而消除了脂多糖的炎癥效應。
磷酸肌醇-4,5-二磷酸3-激酶/蛋白激酶B(PI3K/Akt)信號通路在糖尿病中被抑制。我們的結果表明,干擾TLR4抑制了PI3K/Akt通路,減輕了異常的系膜細胞增殖。本研究證明H?S對高血糖誘導的系膜細胞增殖的保護作用可能與下調PI3K/Akt信號通路有關。更多證據表明PI3K/Akt信號通路參與了H?S的生理效應。
總之,我們發現HG抑制內源性H?S合成并誘導小鼠系膜細胞過度增殖;TLR4激活參與HG誘導的系膜細胞過度增殖;下調TLR4抑制PI3K-Akt通路并抑制小鼠系膜細胞過度增殖。結論是,高葡萄糖可以通過TLR4依賴性方式抑制硫化氫合成誘導小鼠系膜細胞過度增殖。
 相關新聞
相關新聞