熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
2.實驗部分
2.1.試劑
超純水(>18 MΩ.cm)用于膜儲存、納米顆粒合成和脫氯實驗。金(III)三水合物(HAuCl4·3H2O)(Sigma-Aldrich,>99.9%)、鈀(II)氯化物(PdCl2)(Sigma-Aldrich,>99%)、剝離石墨納米片(xGnP)(xG Sciences,Grade M)、乙二醇(EG)(Fluka)、氫氧化鈉(NaOH)(ACS,分析試劑級)、聚砜(PSf)(Udel,P3500)、N-甲基-2-吡咯烷酮(NMP)(Sigma-Aldrich,>99%)、聚乙二醇(PEG 4400)(Sigma-Aldrich,MW=4400 Da)和聚二烯丙基二甲基氯化銨(PDADMAC)(Sigma-Aldrich)按原樣使用。TCE(Sigma Aldrich)、九水合硫化鈉(Sigma Aldrich)和CaCl2(Sigma Aldrich)用于TCE脫氯實驗溶液。
2.2.xGnP負載納米催化劑的合成與表征
制備xGnP固定化填料的方法先前已有報道。簡而言之,制備Au/xGnP納米顆粒,首先制備500 mM的HAuCl4(金鹽前體)水溶液。然后,將50 mg xGnP懸浮在圓底燒瓶中的1 mL PDADMAC和50 mL乙二醇混合物中,并通過超聲處理(Aquasonic 50T浴式超聲儀,VWR Scientific)分散2小時。將所得懸浮液在油浴中加熱至195°C。接著,將50μL金前體溶液和150μL 1 M NaOH加入懸浮液。反應在195°C攪拌下進行30分鐘。制備的Au/xGnP顆粒通過離心從溶液中取出,并用丙酮洗滌三次。制備Pd/xGnP納米顆粒,首先制備22.5 mM的PdCl2(鈀鹽前體)乙二醇溶液。然后,將50 mg xGnP懸浮在18 mL乙二醇中,并通過超聲處理(Aquasonic 50T浴式超聲儀,VWR Scientific)分散12小時。接著,將2 mL 22.5 mM PdCl2溶液加入懸浮液并攪拌2分鐘。攪拌后,溶液在微波(900 W,2450 MHz)中加熱50秒。Pd/xGnP催化劑以與Au/xGnP相同的方式從溶液中分離并洗滌。制備雙金屬Pd-Au/xGnP納米顆粒,首先使用上述Au/xGnP方案將Au沉積在xGnP上。接著,使用與Pd/xGnP方案相同的程序沉積Pd,不同之處在于使用Au/xGnP代替原始xGnP。Pd-Au/xGnP催化劑以與Au/xGnP相同的方式從溶液中分離并洗滌。
通過浸出后使用原子吸收(AA)光譜(Perkin-Elmer 1100)測量Au和Pd的濃度。為了剝離金屬,將稱重量的Au/xGnP、Pd/xGnP和Pd-Au/xGnP在接近沸點(109°C)的王水中加熱。將王水溶液煮沸1小時后,在浴式超聲儀中超聲處理3小時。然后將超聲處理的溶液通過硝化纖維素膜(標稱孔徑0.2μm)過濾,并用水稀釋。使用AA分析稀釋濾液中的Pd和Au含量。
2.3.納米復合中空纖維膜和膜組件的制備
使用pilot-scale中空纖維機器(PHILOS)制造中空纖維膜。所有膜的鑄膜液組成為15 wt%PSf、16 wt%PEG 4400、69 wt%NMP。NMP用作芯液。催化劑填充鑄膜液的納米填料含量為PSf含量的1.25 wt%。鑄膜液和芯液流速分別調整為6.3 mL/min和2.7 mL/min,同時不施加外部溶液。第一和第二凝固浴的溫度均為30°C,牽引速度設定為0.076 m/s。
制備催化劑填充膜時,首先使用浴式超聲將Pd/xGnP或Pd-Au/xGnP懸浮在NMP中。然后,加入PSf和PEG 4400,攪拌24小時直至懸浮液均勻。在中空纖維紡絲前,使用與上述組成相同的純PSf、NMP和PEG 4400溶液清潔PHILOS機器。所有紡絲的中空纖維在水中沖洗24小時,并儲存在水中直至用于脫氯測試。紡絲期間調整的唯一參數是間隙寬度。使用0 cm和2.5 cm的間隙寬度(見補充內容(SC),圖S1)以確定干相轉化對所得膜反應性和滲透性的影響。
制備死端中空纖維組件時,將三根紡絲纖維(直徑=1.4±0.2 mm,長度=300±40 mm)使用環氧樹脂(Loctite Quick Set Epoxy)固定在塑料管中端對端。干燥24小時后,每個組件進行壓力測試檢測泄漏。每個組件中可用于滲透的總纖維外表面積為40.3±10.9 cm2。
2.4.膜表征
2.4.1.膜滲透性
通過記錄在連接到計算機的天平上收集的濾液質量來確定膜通量。所有膜在2.76 bar和22±2°C下壓實,直至達到穩態通量。壓實后,通過滲透通量(L.m-2.h-1)與壓力在0.69-2.76 bar范圍內的線性回歸計算滲透性。
2.4.2.機械性能
使用SII DMS 6100 Exstar進行機械分析確定中空纖維膜的楊氏模量。每3秒以250 N增量進行力測量,共20步。使用中空纖維樣品的橫截面積計算每種膜的楊氏模量。
2.4.3.孔徑測量
使用Quantachrome Porometer 3G(Quantachrome Instruments,USA)量化膜的孔徑分布,能夠測定13 nm至500μm范圍內的孔徑。測量前,每種中空纖維膜用Porofil(Quantachrome Instruments,USA)潤濕。每種中空纖維測試3次。
2.4.4.TEM和SEM成像
對于掃描電子顯微鏡(SEM)成像,通過在中空纖維液氮中冷凍斷裂并濺射金涂層暴露的橫截面來制備膜樣品。樣品使用碳帶安裝在鋁樁上,所有圖像使用FEI Quanta FEG 200 SEM記錄。通過透射電子顯微鏡(TEM)使用JOEL 2200FS顯微鏡表征催化納米顆粒。所有樣品通過將納米顆粒分散在丙酮(約0.01 wt%)中滴鑄在300目鎳或銅網上制備。成像前,網格在90°C干燥24小時。
2.4.5.催化反應性
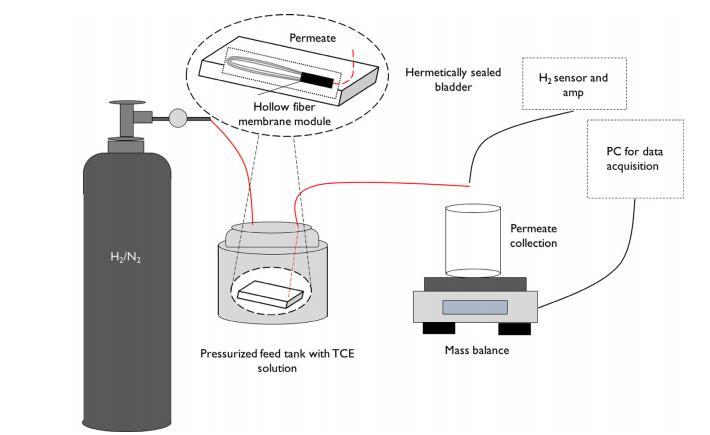
脫氯實驗在流穿模式下進行,使用死端中空纖維組件,在加壓密封氣囊(High Sierra)中零頂空放置于不銹鋼壓力容器中(圖1)。使用微電極和信號放大器(Unisense)監測H2濃度。在6小時泄漏測試中無H2損失。在測量膜反應性前,通過在無反應條件下過濾9.25 mg/L的TCE溶液耗盡吸附容量。耗盡實驗后,使用9.9 mg/L TCE溶液飽和H2(0.8 mM)測定反應性。對于以硫化物作為毒物的實驗,使用氧化處理后的0.7μM或2μM殘留硫化物溶液。中空纖維膜的催化活性按照先前描述的平板膜程序測定。對照測試使用相同進料溶液和相同過濾條件進行,但使用無催化劑的PSf和無催化劑的xGnP/PSf中空纖維膜。
2.4.6.硫化物作為催化劑毒物的影響
使用批次反應器評估硫化物的影響。選擇批次系統是因為它可以直接測定反應常數,而在膜反應器中只能測定反應通量(即觀察到的反應常數kmem和反應器有效長度?eff的乘積)。如果反應器長度已知,可以計算反應常數;然而,反應器幾何形狀以及因此反應器長度僅對最簡單的膜類型(例如徑跡蝕刻膜)有明確定義。如本研究合成的相轉化膜具有復雜的孔空間,使得準確測定kobs mem不可能。
合成硫化物溶液以模擬硫化物地下水通過氯化預處理去除大部分硫化物的過程。通過使用NaOCl氧化0.1 mM硫化物溶液,在0.1 mM CaCl2存在下產生0.2 mM游離氯。選擇CaCl2濃度以代表地下水中可能預期的Ca2+含量。接觸20分鐘后,石膏形成并通過無Pd的xGnP納米復合膜過濾溶液去除。過濾溶液的硫化物濃度使用配備皮安放大器的H2S傳感器測定。為了進行測量,首先將硫化物溶液緩沖至pH 4,以確保所有硫化物物種以H2S形式存在。之后,向溶液中加入9.25 mg/L TCE并用作進料,以測試在殘留硫化物存在下膜的催化活性。
在以硫化物作為催化劑毒物的實驗中,血清瓶完全充滿0.7μM或2μM硫化物溶液,并通過陶瓷濾板通入N2氣體(99.99%純度)鼓泡15分鐘去除溶解氧。去除溶解氧后,將1.25 mg Pd/xGnP或Pd-Au/xGnP納米顆粒加入裝有108 mL溶液的瓶中。然后將所得懸浮液在室溫大氣壓下用H2氣體(99.9%純度)飽和(0.8 mM)15分鐘,并用聚四氟乙烯隔墊和壓蓋密封。注入1 mL 1000 mg/L TCE水溶液開始反應。批次反應器磁力攪拌。從反應器取出的每個樣品通過0.22μm注射器過濾器以去除催化劑并終止反應。通過帶有電子捕獲檢測器的氣相色譜測量TCE還原程度。在氣相色譜圖中,運行期間僅觀察到一個峰。
