熱線:021-66110810,66110819
手機(jī):13564362870
熱線:021-66110810,66110819
手機(jī):13564362870
結(jié)果
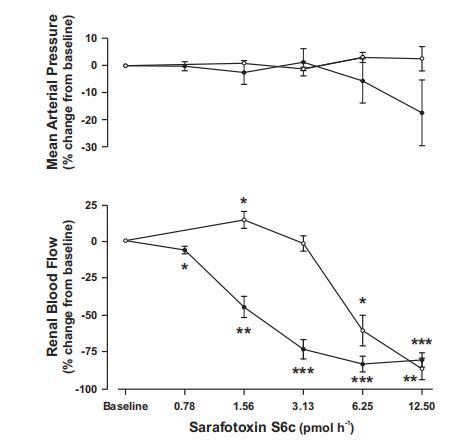
圖1、在正常血糖對(duì)照組(白點(diǎn);n=5)和糖尿病組(黑點(diǎn);n=8)大鼠的腎動(dòng)脈中直接注射增加劑量的內(nèi)皮素B型受體激活劑角蝰毒素6c(S6c)對(duì)平均動(dòng)脈血壓(上圖)和腎血流量(下圖)的急性影響。與組內(nèi)基線相比,*P=0.05、**P=0.01和***P=0.001。
最低劑量的S6c可使對(duì)照組的RBF略有升高,而最高劑量的S6c則可使兩組的血管明顯收縮(圖1)。根據(jù)這些數(shù)據(jù),我們選擇了與0.78pmol/h相對(duì)應(yīng)的劑量,以盡量降低實(shí)驗(yàn)結(jié)束時(shí)腎內(nèi)濃度導(dǎo)致血管收縮的風(fēng)險(xiǎn)。
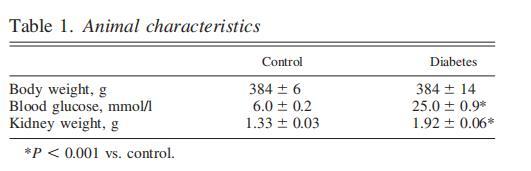
表1、動(dòng)物特征
所有注射鏈脲佐菌素的動(dòng)物都出現(xiàn)了高血糖(表1)。兩組動(dòng)物的體重相似,而糖尿病動(dòng)物的腎臟重量增加(表1)。
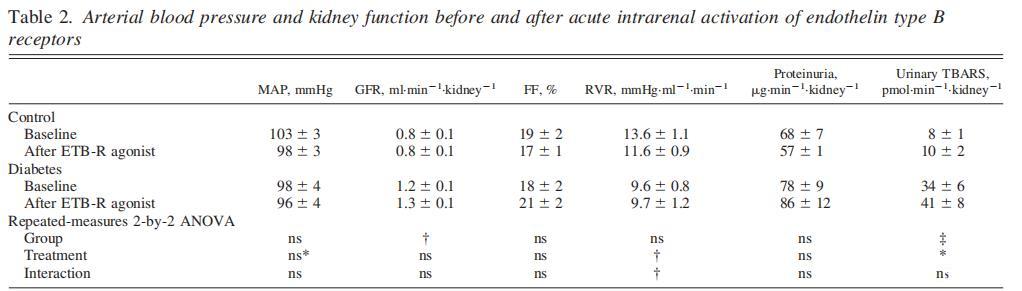
表2、腎內(nèi)B型內(nèi)皮素受體急性激活前后的動(dòng)脈血壓和腎功能
ETB-R激活后,兩組動(dòng)物的血壓均略有下降,糖尿病誘導(dǎo)的腎小球高濾過率不受ETB-R激活的影響(表2)。
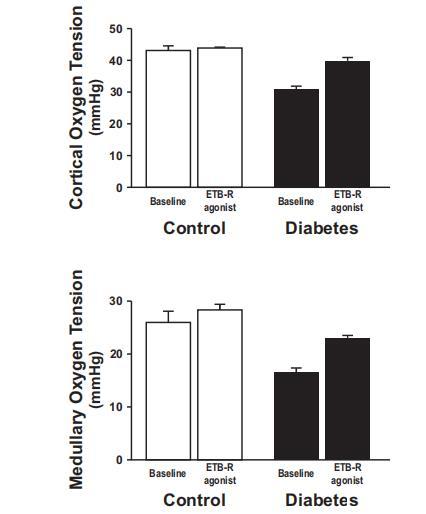
圖2、直接向腎動(dòng)脈注射B型內(nèi)皮素受體(ETB-R)激活劑角蝰毒素6c對(duì)對(duì)照組大鼠(14只)和糖尿病大鼠(9只)皮質(zhì)(上)和髓質(zhì)(下)腎組織氧分壓的影響。糖尿病大鼠腎臟皮質(zhì)和髓質(zhì)組織內(nèi)明顯缺氧,ETB-R激活后明顯改善(圖2)。
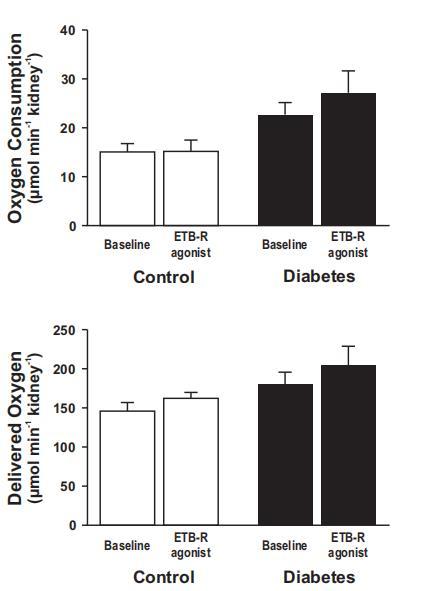
圖3、直接向腎動(dòng)脈注射B型內(nèi)皮素受體(ETB-R)激活劑角蝰毒素6c對(duì)對(duì)照組大鼠(14只)和糖尿病大鼠(9只)腎臟總耗氧量(左圖)和氧輸送量(右圖)的影響。
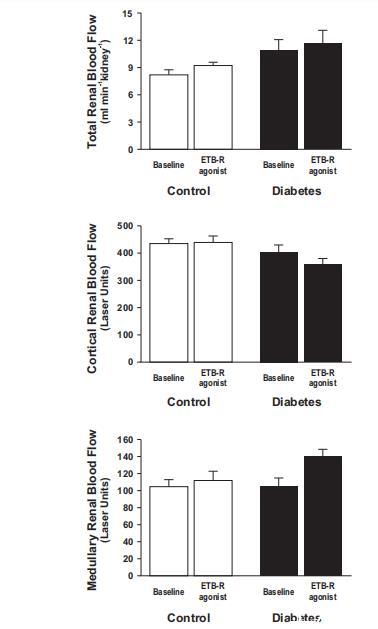
圖4、在對(duì)照組大鼠(n=12-14)和糖尿病大鼠(n=9-12)中,將B型內(nèi)皮素受體(ETB-R)激活劑角蝰毒素6c直接注入腎動(dòng)脈對(duì)腎臟總血流量(左圖)以及皮質(zhì)(中圖)和髓質(zhì)腎血流量(右圖)的影響。
盡管RBF或D˙O2同時(shí)增加,但糖尿病腎臟內(nèi)缺氧是由腎臟總Q˙O2增加引起的(圖3和圖4)。ETB-R激活對(duì)糖尿病誘導(dǎo)的Q˙O2增加沒有顯著影響(圖3)。然而,ETB-R激活后,由于總RBF增加,D˙O2明顯增加(圖3和圖4)。有趣的是,皮質(zhì)和髓質(zhì)RBF受ETB-R激活的影響不同(圖4)。
正如之前所報(bào)道的,糖尿病大鼠尿液中TBARS的排泄量增加(表2)。有趣的是,ETB-R激活會(huì)增加兩組大鼠的尿TBARS。尿蛋白排泄和濾過率均未受影響,而RVR則隨著ETB-R的激活而下降(表2)。
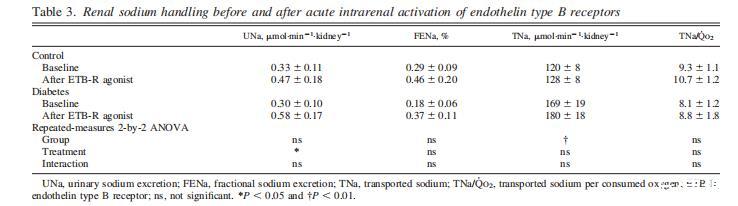
表3、腎內(nèi)B型內(nèi)皮素受體急性激活前后的腎鈉處理
糖尿病大鼠的TNa增加,而與腎臟鈉處理有關(guān)的所有其他參數(shù)在兩組中相似(表3)。ETB-R激活增加了兩組大鼠的尿鈉排泄量。
討論
本研究的主要新發(fā)現(xiàn)是,腎內(nèi)特異性激活ETB-R可通過增加髓質(zhì)RBF和D˙O2快速改善缺氧性糖尿病腎臟的腎內(nèi)PO2。與之前報(bào)道的急性阻斷ETA-R的效果類似,激活ETB-R可改善腎臟PO2,但對(duì)腎臟Q˙O2無重大影響。此外,通過阻斷ETA-R或激活ETB-R來急性調(diào)節(jié)ET受體信號(hào)傳導(dǎo)不會(huì)阻止糖尿病引起的尿蛋白滲漏或氧化應(yīng)激。然而,ETB-R的急性激活會(huì)增加尿鈉排泄。
在糖尿病腎病中,ET-1的過度生成和隨后ETA-R信號(hào)的增加已被證明會(huì)通過誘導(dǎo)內(nèi)質(zhì)網(wǎng)應(yīng)激和細(xì)胞凋亡而導(dǎo)致腎小球和腎小管損傷。ETB-R對(duì)調(diào)節(jié)腎小管鈉和水的處理至關(guān)重要,ETB-R的激活會(huì)抑制腎小管遠(yuǎn)端內(nèi)皮鈉通道對(duì)Na的重吸收。本研究結(jié)果表明,急性激活ETB-R可改善糖尿病腎臟內(nèi)腎組織PO2,這與之前報(bào)道的阻斷ETA-R的效果類似。因此,直接以ETB-R為靶點(diǎn)改善糖尿病腎臟組織PO2并同時(shí)避免使用ETA-R拮抗劑的臨床試驗(yàn)中常見的不必要的液體潴留可能是有益的。事實(shí)上,糖尿病腎臟組織PO2的改善是由髓質(zhì)RBF的增加引起的,這表明ETB-R優(yōu)先參與調(diào)節(jié)髓質(zhì)微循環(huán)。需要注意的是,正如之前所報(bào)道的那樣,腎臟某一部分RBF的改善有可能影響鄰近區(qū)域的氧合。然而,與ETA-R阻斷后的結(jié)果相反,ETB-R刺激對(duì)降低GFR沒有益處。這種對(duì)激活ETB-R和阻斷ETA-R的不同反應(yīng)可能是腎臟微循環(huán)或參與調(diào)節(jié)GFR的結(jié)構(gòu)中不同表達(dá)譜的結(jié)果。
以前曾有報(bào)道稱,糖尿病早期腎內(nèi)缺氧是由導(dǎo)致腎臟Q˙O2增加的幾個(gè)因素造成的。這些因素包括腎小球高濾過導(dǎo)致腎小管電解質(zhì)負(fù)荷增加、腎小管電解質(zhì)轉(zhuǎn)運(yùn)效率降低以及線粒體漏式呼吸增加。除了RBF缺乏代謝控制外,Q˙O2的增加還導(dǎo)致腎內(nèi)缺氧。迄今為止,在胰島素分泌減少的1型糖尿病嚙齒動(dòng)物模型中,使腎內(nèi)組織PO2恢復(fù)正常的大多數(shù)干預(yù)措施也成功地保護(hù)了腎功能并預(yù)防了糖尿病腎病。此外,腎內(nèi)缺氧本身就足以引發(fā)慢性腎病。綜上所述,針對(duì)糖尿病腎臟組織缺氧已成為治療糖尿病腎病的一種可能療法。
S6c是一種強(qiáng)效ET受體激活劑,最早是在蒼術(shù)毒液中發(fā)現(xiàn)的,對(duì)ETB-R的選擇性遠(yuǎn)遠(yuǎn)高于ETA-R。因此,正如之前所報(bào)道的,在最初確定劑量-反應(yīng)關(guān)系的實(shí)驗(yàn)中,S6c濃度越高,血管收縮越強(qiáng),這是血管平滑肌細(xì)胞上的ETB-R被激活的結(jié)果,而不是ETA-R參與作用的結(jié)果。與血管內(nèi)皮表達(dá)的ETB-R相比,血管平滑肌細(xì)胞上的ETB-R對(duì)ET-1的親和力較低,這就解釋了為什么較高濃度的S6c會(huì)導(dǎo)致血管收縮。Schneider等人報(bào)告稱,高鹽飲食喂養(yǎng)大鼠離體傳入動(dòng)脈對(duì)ET-1的血管收縮減弱。有趣的是,抑制ETB-R可阻止對(duì)ET-1的反應(yīng)改變,這凸顯了ET-R信號(hào)改變?cè)谡{(diào)節(jié)RBF中的潛在作用。因此,糖尿病引起的ET受體表達(dá)改變可以解釋本研究中糖尿病腎臟總RBF不受影響的原因。ETB-R激活會(huì)刺激前列腺素的合成,這也可能影響研究結(jié)果。
與慢性治療相比,ETB-R激活對(duì)糖尿病腎功能的急性影響可能在某種程度上有所不同。這也可能解釋了ETB-R激活后對(duì)尿蛋白滲漏明顯缺乏影響以及氧化應(yīng)激顯著增加的原因。事實(shí)上,還需要進(jìn)一步研究確定其對(duì)腎小球?yàn)V過屏障完整性以及腎小管間質(zhì)纖維化發(fā)展的長(zhǎng)期影響,才能使ETB-R激活成為臨床開發(fā)的合適候選藥物。有趣的是,本研究支持使用靶點(diǎn)抑制腎內(nèi)ETA-R信號(hào)或促進(jìn)腎內(nèi)ETB-R信號(hào)以改善整個(gè)腎功能的有益作用。如果是這樣的話,這些結(jié)果將不利于使用非選擇性ET-R拮抗劑治療糖尿病腎病。
總結(jié)
總之,本研究證明急性激活ETB-R可增加糖尿病大鼠腎臟組織的PO2。ETB-R激活后腎內(nèi)PO2的改善主要是髓質(zhì)RBF和D˙O2增加的結(jié)果,而不是對(duì)腎臟Q˙O2的極大影響。本研究結(jié)果表明,ETB-R激活和ETA-R阻斷對(duì)改善糖尿病腎臟組織PO2同樣有效。鑒于使用ETA-R阻斷劑治療慢性腎臟病的初步臨床試驗(yàn)中出現(xiàn)了不必要的體液潴留,因此有必要研究是否可以使用ETB-R激活劑來達(dá)到同樣的腎臟保護(hù)效果,同時(shí)減少嚴(yán)重的副作用。
 相關(guān)新聞
相關(guān)新聞