熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
2.7質譜儀設置
質譜儀LTQ-Orbitrap Velos Pro采用正離子模式。在Orbitrap中進行單次質譜掃描,記錄350至1650m/z之間的質量窗口,最大離子注入時間為250毫秒。分辨率設置為60,000,自動增益控制設置為1,000,000離子。啟用鎖定質量選項后,可通過聚二甲基環硅氧烷背景離子(質子化(Si(CH3)2O))6;m/z 445.120025)對Orbitrap中記錄的光譜進行內部重新校準。實驗采用數據依賴采集模式,交替進行MS和MS/MS實驗。觸發MS/MS的最小MS信號設定為500個離子,使用2 Da的隔離窗口選擇最突出的離子信號進行MS/MS。已被選中用于MS/MS的信號的m/z值被置于排除列表中60秒,排除窗口大小為±10ppm。在所有情況下,記錄一次微掃描。線性離子阱中的CID目標值為5000個離子,最大離子注入時間為150毫秒,歸一化碰撞能量為35%,Q值為0.25,激活時間為10毫秒。每次MS掃描最多觸發20次MS/MS實驗。
2.8數據分析
使用Thermo Scientific Proteome Discoverer軟件1.3版處理原始MS文件。利用MASCOT和SEQUEST搜索引擎對包含正向和反向蛋白質序列的Uniprot人類數據庫中的峰列表文件進行搜索。使用Percolator對搜索到的肽段進行過濾,使其FDR不超過1%,肽段質量偏差設置為10ppm,并且要求每個識別出的肽段至少包含6個氨基酸。數據庫搜索參數為前體質量容差20ppm,片段質量容差CID 0.8 Da,動態修飾為脫氨基(N、Q)、氧化(M),靜態修飾為IAM烷基化(C)。在所有搜索中,都選擇了胰蛋白酶進行兩次漏切。蛋白質的分組采用了最大解析規則。用于確定蛋白質絕對定量的計算方法是取含量最高的三個肽段的平均值。
2.9統計和生物信息學分析
導出蛋白質特征和定量數據,用于進一步的統計和生物信息分析。每個無標簽定量數據都按相應的尿量進行劃分,并采用量化法進行歸一化處理。歸一化后,使用主成分分析(PCA)和斯皮爾曼相關性等無監督分層聚類分析來識別離群值和樣本相似性。采用非參數曼-惠特尼U檢驗法檢測外泌體和尿瘤樣本之間的差異表達蛋白。使用本杰明-霍奇伯格假發現率(Benjamini-Hochberg false discovery rate)對P值進行多重檢驗調整。如果調整后的P值為0.05,且fold change<-2或>2,則認為蛋白質在兩種情況下有顯著差異表達。此外,蛋白質必須在兩個隊列中至少有50%的樣本中被發現。火山圖用于快速顯示統計差異。不同樣本蛋白質組的分層聚類分析結果通過熱圖的平均值顯示。所有統計分析均使用R軟件包進行。
Cytoscape軟件用于基因本體和通路分析。根據從UniProt數據庫中提取的基因本體注釋,提交了外泌體和尿瘤間差異富集蛋白的統計列表,以確定外泌體囊泡中生物過程的富集情況。
2.10耗氧量和ATP合成測量
氧氣消耗量是在配備有安培電極的恒溫氧儀(Unisense微呼吸系統,UnisenseA/S,丹麥)上測定的。每次實驗前用培養基平衡電極,并生成標準曲線。培養基包含120mM KCl、2mM MgCl2、1mM KH2PO4、50mM Tris-HCl、pH7.4和25μg/mL氨芐西林(最終體積1.7mL,溫度23℃)。呼吸速率以nmol O/min/mg表示。
ATP合成采用熒光素/熒光素酶化學發光法測定,如報告所述。簡而言之:樣品首先在37℃下孵育10分鐘:100mM Tris/HCl(pH7.4)、100mM KCl、1mM EGTA、2.5mM EDTA、5mM MgCl2、0.2mM二(腺苷-5′)五磷酸鹽、0.6mM歐貝因、氨芐青霉素(25μg/mL)、5mM KH2PO4。為誘導ATP合成,在相同pH值的混合物中加入0.1mM ADP。然后用熒光素/熒光素酶化學發光法在發光儀中測量ATP濃度,ATP標準溶液在10-9和10-5M之間,用于校準。為了刺激由復合物I+III+IV組成的通路,氧消耗和ATP合成分析均使用了0.7mM NADH。為了刺激由復合物II+III+IV組成的途徑,使用了20mM丁二酸。
3結果
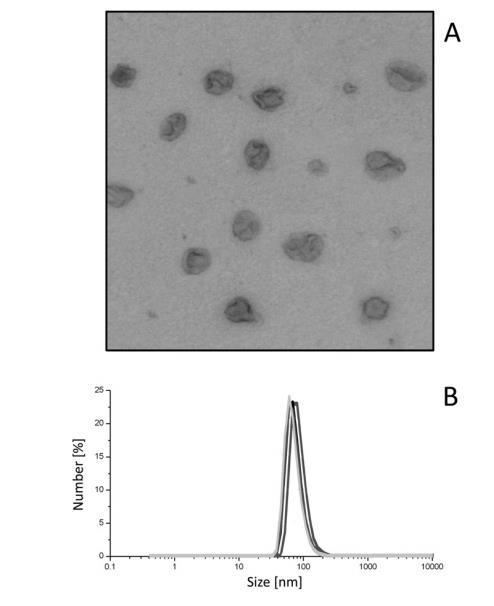
圖1分離的外泌體的特征。圖A:分離的尿液外泌體的代表性透射電子顯微鏡圖像。圖B:通過動態光散射評估的外泌體大小分布圖。該圖顯示了一個高斯分布曲線,平均峰值為80±8nm。
從6名男性和6名女性健康供體的第二次晨尿樣本中獲取外泌體和尿瘤。外泌體通過超速離心法分離。為確定其大小和形態,進行了透射電子顯微鏡(TEM)和動態光散射分析。圖1A顯示,超速離心得到的餾分由顯示典型外泌體杯狀形態的囊泡組成。通過動態光散射分析確定了囊泡的大小,結果顯示囊泡呈高斯分布,平均峰值為80±8nm(圖1B)。
采用兩種不同的方法對獲得的樣本進行尿瘤特征測定。第一種方法是凍干100微克的等分樣品,然后送去質譜分析(未經處理的部分)。第二種方法是在質譜分析之前,用有機溶劑處理相同數量的樣品(100微克),以富集疏水性較強的蛋白質(沉淀部分),并用組合肽配體庫珠子(CPLLbs)處理親水相,以富集低豐度蛋白質(珠子和未結合部分)。
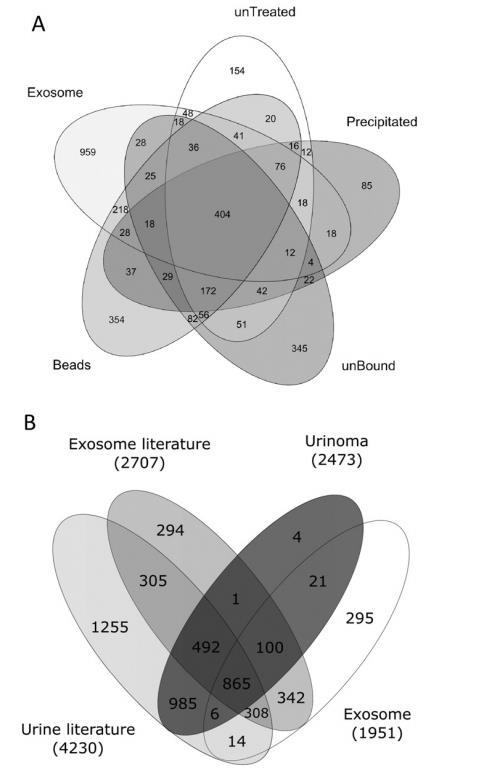
圖2維恩圖。A:外泌體和尿液中總蛋白質的維恩圖;B:文獻和我們的數據以及健康供體尿瘤中外泌體數據的維恩圖,顯示常見和不常見的蛋白質。數字代表各自重疊和非重疊區域中的不同蛋白質。
在外泌體、未處理、沉淀、珠子和未結合餾分中分別鑒定出1951、1176、993、1614和1347個蛋白質。各部分鑒定出的蛋白質數量相似。在尿瘤(共2469個蛋白質)中,珠狀體(14%)、未結合(14%)和沉淀(3%)部分分別鑒定出354個獨有蛋白質、345個獨有蛋白質和85個獨有蛋白質。相比之下,未經處理的樣品中只發現了154個專屬蛋白質(6%)。這證實了在質譜分析前進行樣品分餾的優勢,可以在蛋白質濃度動態范圍較大的生物樣品中鑒定出大量蛋白質。圖2A顯示了健康供體的外泌體和尿瘤蛋白質之間的重疊程度。共有3429個蛋白質存在,其中只有992個(29%)與外泌體和尿瘤相匹配。外泌體中的其余959個蛋白質(28%)和尿瘤中的1478個蛋白質(43%)不屬于這兩組中的一組。
12個生物重復序列的平均變異系數為0.5,各個蛋白質組之間沒有發現任何關系。例如,它們沒有因性別而聚類。此外,通過使用DellR.B報告的公式,我們可以得出結論:12個生物重復樣本足以鑒定出兩倍變化的定量蛋白質變異特征,顯著性水平為0.05,功率為80%。
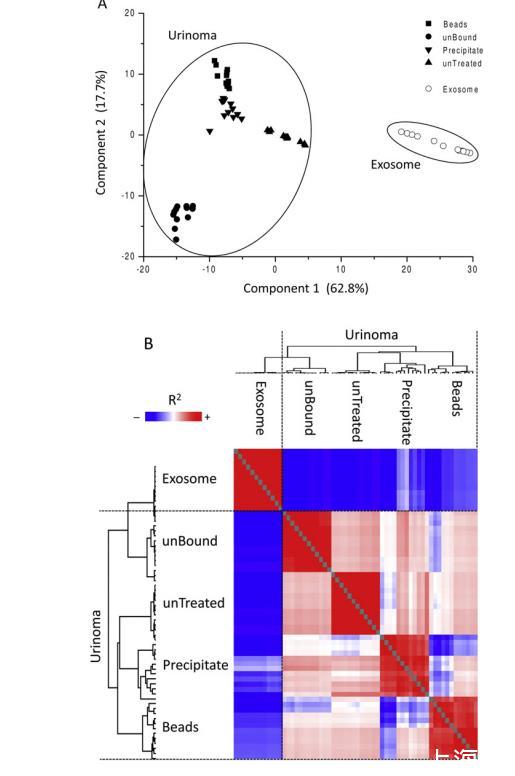
圖3與外泌體和不同尿液組分的無監督分層聚類分析相關的主成分分析和斯皮爾曼相關圖。樣本來自21名健康捐獻者。A,外泌體(圓圈)和尿液組分主成分分析的二維散點圖,即:未處理(上三角)、沉淀(下三角)、珠子(正方形)和未結合(點)。B,斯皮爾曼相關圖:系數值用從0.1(藍色)到0.9(紅色)的偽彩色標尺表示。此外,樹狀樹枝圖顯示了無監督分層聚類分析的結果,將相似的斯皮爾曼系數值相互靠近。兩幅圖都顯示了外泌體和尿瘤之間兩個明顯的獨立聚類。
對不同的尿液組分和外泌體進行了主成分和層次聚類分析,以突出兩組樣本的異同。外泌體和其他尿液組分的主成分二維散點圖顯示,在所有樣本中,外泌體和尿瘤的蛋白質都有很好的分離(圖3A)。根據蛋白質組的豐度特征,使用與分層聚類分析相關的斯皮爾曼相關圖也得到了相同的結果(圖3B)。此外,不同尿液組分的平均斯皮爾曼系數(R2)(0.74±0.11)高于外泌體和尿液組分之間的平均斯皮爾曼系數(0.26±0.08)。這證實外泌體和尿瘤組是兩個獨立的群組。
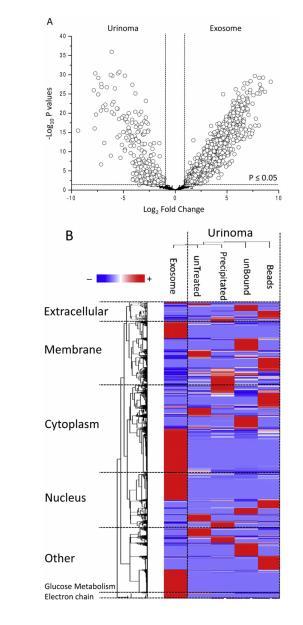
圖4外泌體和尿瘤分層聚類分析的火山圖和熱圖。A,根據外泌體和尿瘤中所有已鑒定蛋白質的折疊變化(Log2)和P值(-Log10)繪制的火山圖。白色圓圈表示外泌體和尿瘤中發生顯著統計學變化的1660個蛋白質。共有1464個蛋白質在外泌體中富集(右側),而196個蛋白質在外泌體中代表性不足(左側)。P值為>0.05的蛋白質顯示為黑點。B,基于細胞組分基因本體注釋的外泌體和不同尿液組分蛋白質組的無監督二維分層聚類熱圖。在熱圖中,每一行代表一種蛋白質,每一列對應每組12個樣本的平均值。與每個蛋白質的平均值相比,蛋白質豐度的歸一化Z評分用偽彩色標度表示,正表達(紅色)、等表達(白色)和負表達(藍色)。樹狀樹枝圖顯示了無監督分層聚類分析的結果,將相似的蛋白質組特征值相互靠近。對樹枝圖和熱圖的目視檢查表明,外泌體和尿瘤組有明顯的聚類。
為了更好地描述這兩組之間的差異,我們根據細胞成分基因本體注釋,對蛋白質豐度進行了火山圖和熱圖分層聚類分析。火山圖顯示,與尿瘤相比,外泌體中共有1660個蛋白質發生了顯著變化。其中,1464個蛋白質在外泌體中富集(圖4A)。蛋白質組譜熱圖顯示,細胞質(34%)和細胞膜(21%)區明顯富集的蛋白質占多數,而核區(13%),特別是細胞外區(1%)與蛋白質組總量相比代表性不足。研究發現,葡萄糖代謝和呼吸鏈等代謝途徑的蛋白質含量明顯增加。特別是最后兩種途徑,在這里首次得到了強調。
 相關新聞
相關新聞