熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
5.siRNA-GPR4可降低酸性微環境中EPC的血管生成能力
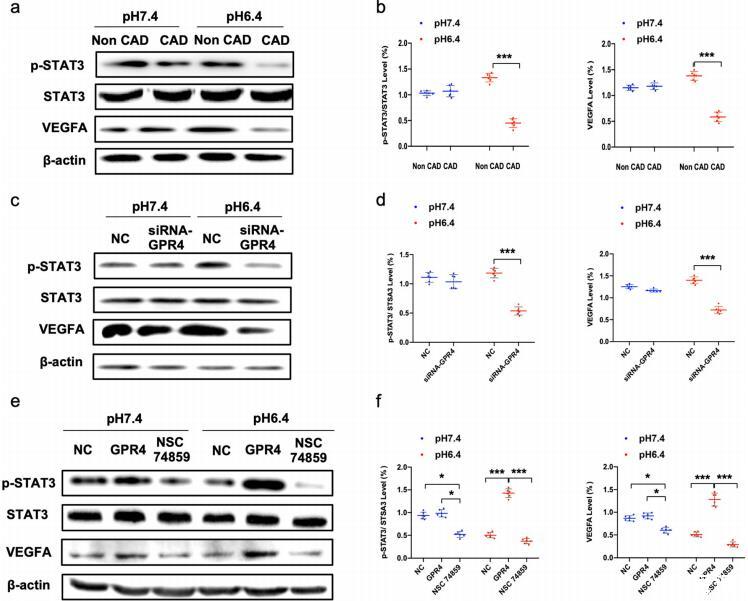
圖5:GPR4水平的變化影響酸介導的EPCs血管生成。a,b不同pH環境下非CAD和CAD患者EPCspSTAT3、STAT3和VEGFA蛋白表達的代表性照片和定量分析。c-f不同pH環境下非CAD和CAD患者EPCs中p-STAT3、STAT3和VEGFA蛋白表達的代表性照片和定量分析。用siRNA-GPR4和陰性對照siRNA(NC)或表達GPR4的質粒、陰性對照質粒(NC)和NSC74589轉染EPC。
此外,我們還研究了GPR4信號通路激活對STAT3磷酸化(p-STAT3)和VEGFA表達的影響。我們的數據顯示,與非CAD患者相比,p-STAT3和VEGFA的表達在pH值為6.4時明顯降低,而在pH值為7.4時組間差異不顯著(圖5a,b)。為了進一步證實GPR4在酸性微環境中的血管生成能力,我們用siRNA沉默了EPCs中GPR4的表達。免疫印跡證實了GPR4、STAT3和VEGFA在EPCs中的表達。我們的數據顯示,siRNA-GPR4在pH6.4時會降低p-STAT3和VEGFA的表達,但在pH7.4時不會(圖5c,d)。
6.STAT3抑制劑可阻斷GPR4上調后體外和體內血管生成的增加
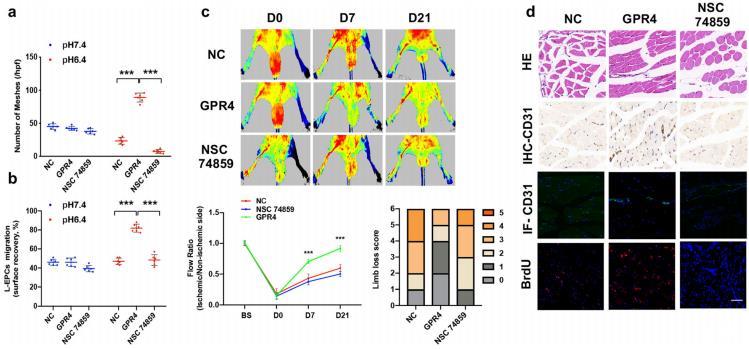
圖6:GPR4在酸性微環境的刺激下通過STAT3信號通路調節來自CAD患者的EPCs中VEGFA的表達。a,b在不同pH環境的EGM培養基中培養6h后定量分析管形成(a)和遷移(b)。c注射轉染了陰性對照質粒(NC)或表達GPR4和NSC74859的質粒的EPC后,裸鼠后肢缺血血流的代表性照片和定量分析。d缺血后肢的HE、IHC-CD31、IF-CD31和BrdU染色切片。
我們的數據表明,STAT3抑制劑NSC74859可以降低GPR4基因轉移上調的VEGFA水平(圖5e、f),這一發現也與遷移和基質膠實驗結果平行(圖6a、b)。此外,NSC74859還消除了GPR4基因轉染引起的裸鼠后肢皮下血流增加(圖6c)。后肢的免疫組化和免疫熒光見圖6d。
討論:
我們目前的研究發現,與健康人相比,CAD患者體內GPR4的表達明顯減少,這導致EPCs在酸性缺血環境中的血管生成能力受損。GPR4表達的上調明顯提高了CAD患者體內和體外酸性微環境中EPCs的適應性血管生成能力。從機理上講,GPR4表達的增加可增強H+的結合,導致下游STAT3/VEGFA信號通路的表達增加,最終加速EPCs的血管生成。GPR4可能是EPCs適應酸中毒環境和恢復受損血管生成能力的一個有前途的靶點。
人們普遍認為,EPC是血管修復和再生醫學中移植治療的重要組成部分。盡管基于EPC的CAD實驗研究正在廣泛展開,但盡管一些研究觀察到功能改善,但仍存在爭議。其中,來自CAD患者的EPC數量較少且功能受損。因此,探索EPCs在酸性條件下的治療模式及其內在機制至關重要。
酸性微環境作為缺血的病理生理化學因素,可能會顯著影響動員的EPCs的功能。有趣的是,以前的數據表明,EPCs暴露于酸性介質對細胞功能有雙重影響。人們普遍認為,酸性微環境會對干細胞/祖細胞產生額外的破壞作用,因為細胞外pH值的降低會誘導細胞酸化,從而導致細胞死亡。另一方面,越來越多的數據顯示,酸性環境也能提供增強細胞能力的刺激,這表明嚴峻的微環境能激發細胞自我適應/防御,以保持其能力。值得注意的是,有證據表明,在適當的時候,酸性微環境可以促進健康人的EPCs血管化。
越來越多的數據表明,H+可引發微環境對各種細胞(如癌細胞和內皮細胞)的影響,質子感應GPCR被認為是介導下游細胞內信號通路的相關受體。Wyder等人的研究表明,與野生型小鼠相比,缺乏GPR4的小鼠的腫瘤生長、細胞增殖以及血管形態、長度和密度的改變都嚴重減少。鑒于質子感應GPCR與酸中毒之間的關系,質子感應GPCR家族成員可能在EPCs中發揮作用,并在CAD患者的EPCs中退化。有趣的是,Ding等人發現質子感覺蛋白OGR1通過酸激活介導了對EPCs增殖和血管生成的抑制作用。在本研究中,我們評估了EPCs中質子傳感GPCR的水平,與OGR1、TDAG8和G2A相比,GPR4的表達相對豐富,這表明EPCs可能通過GPR4感知局部酸性微環境,以調整自身適應破壞性環境。此外,與健康受試者相比,CAD患者的EPC中GPR4的表達明顯減少,這與酸性誘導的新生血管生成受損相一致。因此,EPCs的GPR4表達和血管生成能力的平行趨勢表明,GPR4可能介導了酸性微環境中EPCs細胞功能的異常。我們的研究結果進一步證明,EPC中可能存在各種質子感應GPCR,它們在酸性環境中可能會相互影響。
據報道,GPR4在腫瘤和血管內皮細胞中過度表達,可誘導血管生成增加。有趣的是,在一些研究中,GPR4的過表達也呈現出負的細胞活性,這表明GPR4對不同類型細胞的影響是有爭議的。Kim等人報道,SPC/GPR4相互作用誘導體內EC的血管生成,磷脂酰肌醇-3激酶和Akt的激活均參與其中,這表明GPR4可能是EC血管生成能力的關鍵調節因子。Ren等人進一步揭示,GPR4的過表達介導了Notch1信號的細胞延伸,而這一增量與體外HMEC-1細胞內皮管形成的增加相關。因此,GPR4可作為內皮細胞系血管穩態的靶點。為了進一步證明GPR4在EPC介導的CAD患者血管生成能力中的作用,我們通過pcDNA3.1(+)-GPR4提高了GPR4的表達,并刺激EPC對酸性刺激做出反應。事實上,我們的研究結果表明,與pH值為7.4的正常微環境相比,pcDNA3.1(+)-GPR4基因轉移可促進pH值為6.4的CAD患者EPC的血管生成。與此相一致,我們的數據顯示,在體外和體內pH值為6.4時,GPR4表達的增加與EPC粘附、遷移和管道形成能力的增加是平行的。此外,siRNA-GPR4基因轉移誘導健康受試者的EPC中GPR4表達減少,EPC的體內外能力在pH6.4時也出現下調。然而,在pH值為7.4時,EPCs并未出現上述功能變化。因此,我們目前的研究表明,GPR4表達的降低至少部分導致了酸性環境下CAD中EPC介導的血管生成下降,為我們理解酸性微環境中的EPC血管生成功能障礙的分子機制提供了新的見解。
轉錄信號轉導和激活因子(STAT)家族是細胞質轉錄因子,可促進細胞內信號通路對細胞因子和因子的反應。它由七個成員組成:STAT1、STAT2、STAT3、STAT4、STAT5A、STAT5B和STAT6。STAT3被認為能促進細胞的生長和存活、血管生成、遷移、侵襲或轉移,有報道稱它受GPCRs的調控。先前的研究表明,在酸性腫瘤環境中,通過抑制STAT3VEGF在體外和體內的表達,可減輕乳腺癌細胞的血管生成和腫瘤生長。此外,STAT3誘導的血管生成也在其他類型的細胞中得到證實,如肝細胞癌。因此,我們提出了一個假設,即STAT3可能是與酸有關的EPCs血管生成信號通路的重要步驟之一。然而,STAT3是否能激活酸介導的新生血管形成仍是未知數。在我們的研究中,我們發現轉染GPR4提高了CAD患者EPCs的血管生成能力并增加了VEGFA的表達,而敲除GPR4則抑制了血管生成能力。此外,我們的數據還顯示,p-STAT3/STAT3的比值增加,這與VEGFA表達的增加和GPR4表達的改變相一致。這些結果表明,GPR4介導的酸誘導血管生成與STAT3/VEGFA通路之間可能存在相關性。此外,當STAT3被阻斷時,酸性微環境介導的血管生成中增加的體外和體內EPCs功能受到抑制。這些結果表明,GPR4/STAT3/VEGFA信號通路參與了酸性微環境中EPC的功能調節。
然而,我們目前的研究還存在一些局限性。首先,酸性微環境是一個由多種質子感應蛋白組成的復雜微系統。GPR4信號與其他受體的聯系還需要進一步研究。此外,與CAD發病率和預后相關的GPR4信號通路也值得闡明。
結論:
總之,本研究首次證明了GPR4/STAT3/VEGFA信號通路導致EPCs血管生成能力受損,而提高GPR4可恢復CAD患者EPCs的血管生成能力。這些發現為治療CAD提供了新的細胞治療靶點。
 相關新聞
相關新聞