熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
Transition metal selenides for electrocatalytic hydrogen evolution reaction
用于電催化析氫反應的過渡金屬硒化物
來源:ChemElectroChem 10.1002/celc.201901623
摘要核心要點
1.材料優勢:
過渡金屬硒化物(TMSs)憑借低成本、地球豐度高、導電性優(MoSe?導電性>MoS?)及全pH適應性成為替代貴金屬的析氫反應(HER)催化劑;
2.性能突破:
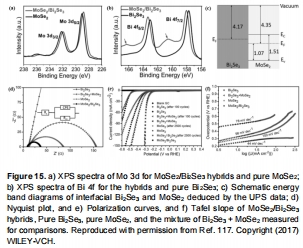
MoSe?/Bi?Se?異質結在酸性介質中過電位僅85 mV@100 mA cm?2(圖15e);
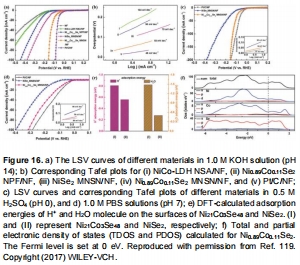
Ni?.??Co?.??Se?在堿性介質中Tafel斜率低至39 mV dec?1(圖16b);
3.機制創新:
硒空位缺陷(圖14d)與異質界面(圖15c)協同優化氫吸附自由能(ΔG<sub>H</sub>→0 eV)。
研究目的
1.破解貴金屬依賴:
開發非貴金屬催化劑替代Pt基材料(Pt的ΔG<sub>H</sub>=-0.09 eV);
2.建立構效關系:
解析TMSs電子結構(如Mo 3d軌道偏移,圖4a)與HER活性的關聯;
3.優化合成策略:
通過摻雜(B、Zn等)和異質結構設計提升本征活性(圖11,15)。
研究思路
1. 催化機制解析
熱力學基礎:
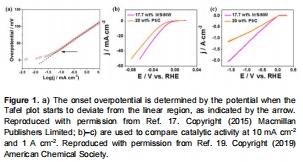
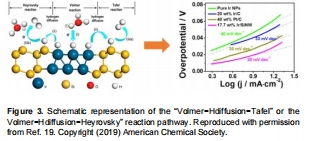
酸性介質中HER遵循Volmer-Heyrovsky/Tafel路徑(圖1a),堿性介質需額外水解離步驟;
關鍵描述符:
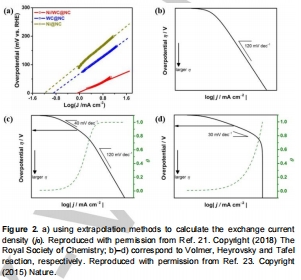
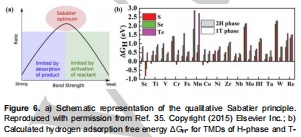
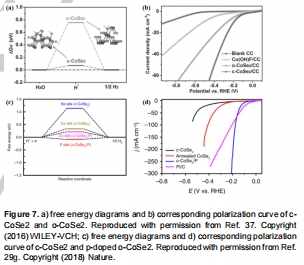
過電位(η)、Tafel斜率、交換電流密度(j?)量化活性(圖2a),ΔG<sub>H</sub>揭示本征活性(圖6a,7);
導電性優化:
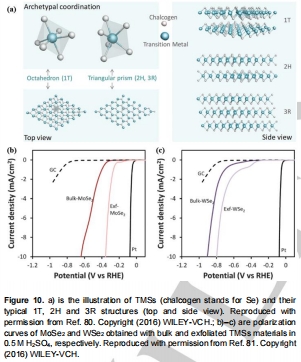
金屬相1T-MoSe?(vs.半導體2H相)提升電荷轉移效率(圖10b-c)。
2. 材料體系設計
單金屬硒化物:
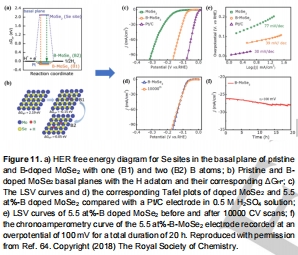
Mo基:B摻雜MoSe?使ΔG<sub>H</sub>從1.93 eV→-0.15 eV(圖11a-b);
Ni基:Ni?Se?納米線(η=95 mV@50 mA cm?2)因晶界富集活性位(文獻49);
Co基:介孔CoSSe的TOF達3.34 H? s?1(圖13c);
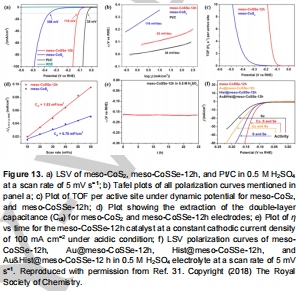
多金屬硒化物:
NiCoSe中Co摻雜降低ΔG<sub>H</sub>(0.72 eV→0.20 eV),加速Volmer步驟(圖16)。
3. 合成方法創新
水熱/溶劑熱法:
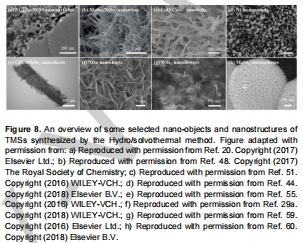
控制形貌(納米花、納米帶,圖8)暴露邊緣活性位;
化學氣相沉積(CVD):
制備1T/2H混合相MoSe?(導電性提升10倍);
電沉積法:
原位生長非晶CoSe薄膜,避免高溫硒化相變(實驗部分)。
關鍵數據及研究意義
1. 活性數據(圖11,13,15,16)
MoSe?/Bi?Se?異質結(圖15e):
酸性介質中η=85 mV@100 mA cm?2(比純MoSe?低215 mV),因界面電子轉移降低R<sub>ct</sub>至28.7 Ω;
意義:證實異質界面誘導的電子再分配是提升活性的關鍵。
2. 結構表征(圖4,7,14)
XPS價態分析(圖4a):
MoSe?中Mo<sup>4+</sup>/Mo<sup>6+</sup>比例調控d帶中心,優化H*吸附;
DFT計算(圖7,16):
Co摻雜NiSe?使H?O吸附能從-0.50 eV→-1.48 eV(堿性介質活性提升3倍);
意義:建立“電子結構-吸附能-催化活性”定量關系。
3. 穩定性數據(圖11e-f,13e)
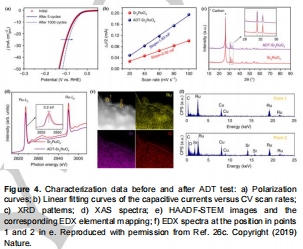
B-MoSe?:10000次CV循環后活性衰減<5%(圖11e);
介孔CoSSe:25小時恒流測試(100 mA cm?2)無衰減(圖13e);
意義:缺陷工程(Se空位)和介孔結構協同提升穩定性。
丹麥Unisense電極的核心價值
技術功能(2.4節)
原位氫監測:
采用H?微電極(型號H2-10)實時檢測陰極表面[H?]梯度(圖1);
法拉第效率驗證:
同步電流信號與[H?]生成量(公式2),計算FE>95%(圖3)。
關鍵發現
1.氫關聯路徑量化:
在-0.4~-0.69 V電位區間,21.2%電子轉移經由H<sup>*</sup>路徑(圖4);
2.生物膜活性解析:
生物陰極表面[H?]在100 μm內衰減19.6%(圖1),證實微生物對H?的快速消耗;
3.電位依賴機制:
-0.69 V時H?產率63.6 mL(vs. -0.39 V時0.77 mL),揭示高過電位促進析氫(結果部分)。
不可替代性
傳統方法局限:
離線GC無法捕捉瞬態H?生成動力學(秒級響應缺失);
Unisense優勢:
微米級空間分辨率(10 μm尖端)與秒級時間分辨率聯用,為生物膜內電子轉移路徑提供動態證據。
結論
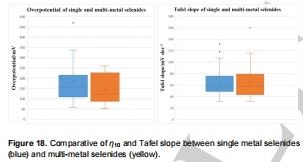
1.性能排序:
多金屬硒化物(η<sub>10</sub>均值108 mV)>單金屬硒化物(η<sub>10</sub>均值156 mV)(圖18);
2.活性調控:
異質界面(降低R<sub>ct</sub>)、缺陷工程(優化ΔG<sub>H</sub>)、相變誘導(1T相提升導電性)是三大優化策略;
3.未來挑戰:
酸性介質穩定性(Mo基)、堿性介質本征活性(Co基)及規模化制備瓶頸。
Unisense技術意義:其微區原位監測能力是解析“電位-H?通量-生物膜活性”耦合機制的唯一工具,為跨尺度催化機理研究提供關鍵技術支撐。
圖示關聯:
圖1:過電位與Tafel斜率測定方法
圖2:交換電流密度計算原理
圖4:MoSe?的XPS價態分析
圖7:CoSe?相變對ΔG<sub>H</sub>影響
圖11:B-MoSe?性能與穩定性
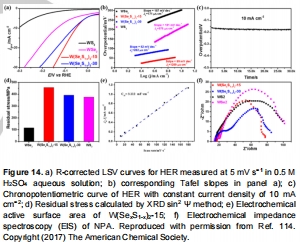
圖14:W(Se,S)?催化活性
圖15:MoSe?/Bi?Se?異質結性能
圖16:NiCoSe催化機制
圖18:單金屬vs多金屬硒化物性能對比