熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
The role of adipose tissue and subsequent liver tissue hypoxia in obesity and early stage metabolic dysfunction associated steatotic liver disease
脂肪組織和隨后的肝組織缺氧在肥胖和早期代謝功能障礙相關脂肪性肝病中的作用
來源:International Journal of Obesity (2024) 48:512–522 (DOI: 10.1038/s41366-023-01443-w)
摘要內容
本研究通過高脂高果糖飲食(HFHFD)誘導小鼠肥胖模型,探究進行性肥胖對脂肪組織和肝臟缺氧的影響。主要發現包括:
脂肪組織缺氧:在肥胖早期(8周)即出現,并隨肥胖進展持續惡化。
肝臟缺氧:僅在肥胖后期(16–20周)出現,此時小鼠處于代謝功能障礙相關脂肪性肝病(MASLD)的早期階段(單純脂肪變性)。
模型對比:在膽堿缺乏高脂飲食(CDAHFD)的非肥胖MASLD模型中,肝臟缺氧未顯著出現,表明肥胖相關過程(如脂肪組織缺氧)是肝臟缺氧的關鍵驅動因素。
分子驗證:缺氧相關基因(如Slc2a1、Ldha)表達變化與直接氧分壓測量一致,證實缺氧在肥胖和MASLD病理中的作用。
研究目的
本研究旨在:
揭示進行性肥胖中脂肪組織和肝臟缺氧的動態演變時序。
探究缺氧與代謝紊亂(如胰島素抵抗、血脂異常)的關聯及其在MASLD早期發病機制中的作用。
比較肥胖相關MASLD(HFHFD模型)與非肥胖MASLD(CDAHFD模型)的缺氧差異,驗證肥胖在缺氧發展中的必要性。
研究思路
研究采用兩步法實驗設計:
HFHFD肥胖模型:8周齡雄性C57BL/6J小鼠喂養高脂高果糖飲食4、8、12、16、20周,模擬進行性肥胖和早期MASLD。
CDAHFD非肥胖模型:小鼠喂養膽堿缺乏高脂飲食1–6周,誘導嚴重脂肪性肝炎(MASH)但不伴肥胖,用于驗證肝臟缺氧的肥胖依賴性。
關鍵方法包括:
直接缺氧測量:使用Unisense微電極活體檢測組織氧分壓(pO?)。
間接缺氧評估:缺氧探針(pimonidazole)染色、缺氧相關基因PCR陣列。
多維度表型分析:體重、血清生化(ALT、AST、血脂)、葡萄糖/胰島素耐受測試、肝臟病理(NAS評分)、脂肪細胞肥大分析。
統計分析:混合效應模型評估組間差異,Spearman相關性分析缺氧與代謝參數的關聯,建立核心缺氧基因特征模型。
測量數據及研究意義
體重與代謝參數
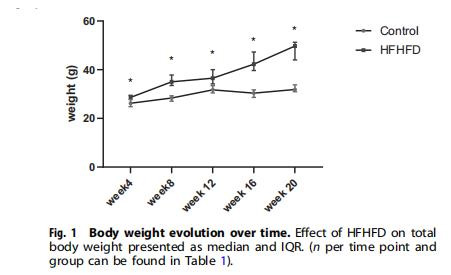
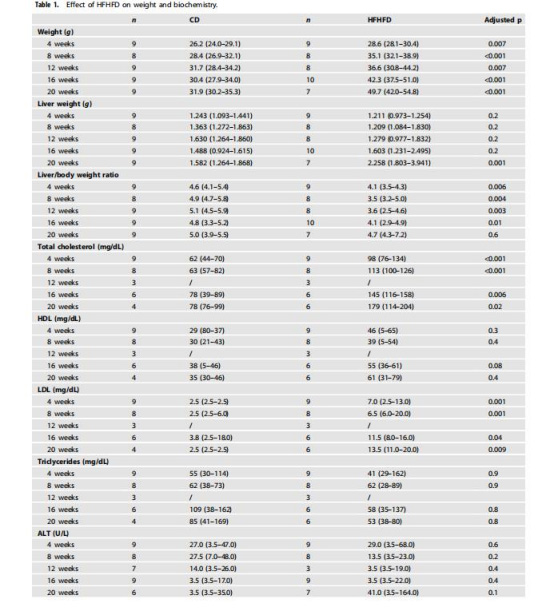
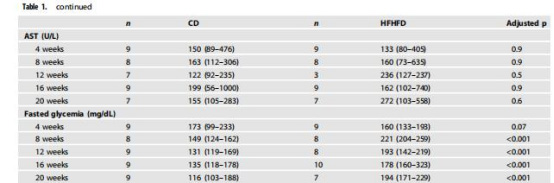 數據來源:表1(體重、血脂、肝酶等)、圖1(體重動態變化)。
數據來源:表1(體重、血脂、肝酶等)、圖1(體重動態變化)。
研究意義:證實HFHFD誘導進行性肥胖、血脂異常(總膽固醇↑、LDL↑)和胰島素抵抗,提供模型有效性基礎;肥胖程度與缺氧發展正相關,提示代謝紊亂是缺氧的驅動因素。
組織病理學評估
數據來源:圖2(肝臟NAS評分和H&E染色)、(脂肪細胞直徑)。
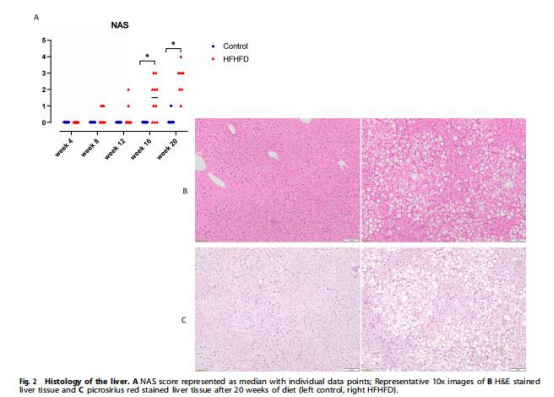
研究意義:肝臟NAS評分↑顯示單純脂肪變性(早期MASLD),但無纖維化;脂肪細胞直徑↑表明脂肪組織擴張,解釋缺氧源于灌注不足;病理變化與缺氧測量協同驗證模型生理相關性。
活體組織氧分壓(pO?)
數據來源:圖3(性腺脂肪和肝臟pO?動態)。
研究意義:直接量化缺氧程度:性腺脂肪pO?從8周顯著↓(對照組~40 mmHg → HFHFD 8周↓~18.9 mmHg),肝臟pO?從16周↓(對照組~30 mmHg → HFHFD 16周↓~15 mmHg);CDAHFD模型無顯著pO?↓(圖3C),證明肝臟缺氧依賴肥胖相關代謝紊亂。
缺氧探針染色與基因表達
數據來源:圖5–6(基因表達)、表4(核心缺氧基因)。

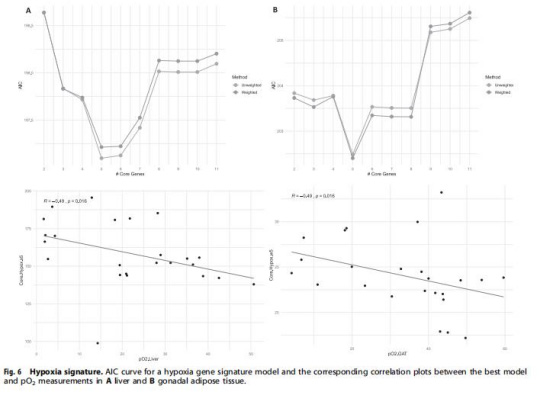
研究意義:pimonidazole染色面積↑與pO?↓相關(r=-0.27),驗證直接測量可靠性;基因表達(如脂肪組織Slc2a1↑、肝臟Ldha↑)揭示缺氧驅動糖酵解增強(Warburg效應);建立組織特異性缺氧基因特征(脂肪:Eno1、Mif等;肝臟:Ldha、Slc2a1等),為缺氧提供分子標志物。
缺氧-代謝參數相關性
數據來源:表3(Spearman相關性)。
研究意義:脂肪pO?與血糖(r=-0.61)、LDL(r=-0.45)顯著負相關,表明缺氧直接促進代謝紊亂;肝臟pO?與LDL負相關(r=-0.53),支持缺氧在MASLD進展中的因果作用。
結論
缺氧時序性:脂肪組織缺氧是肥胖早期事件(始于8周),肝臟缺氧發生于肥胖后期(16–20周),此時處于MASLD早期(單純脂肪變性)。
肥胖的必要性:非肥胖CDAHFD模型雖誘導嚴重MASH,但無顯著肝臟缺氧,表明肥胖相關過程(如脂肪組織缺氧驅動的脂解↑和肝脂質沉積)是肝臟缺氧的前提。
病理機制:缺氧通過激活HIF-1α等通路,改變代謝/血管生成基因表達(如Slc2a1↑促進葡萄糖攝取),加劇胰島素抵抗和肝脂肪變性。
臨床啟示:靶向脂肪組織缺氧或成預防MASLD的新策略(如HIF抑制劑);直接pO?測量為缺氧診斷提供金標準。
丹麥Unisense電極測量數據的詳細解讀
測量方法概述:
技術細節:使用Unisense Clark型微電極(尖端直徑25 μm),活體插入性腺脂肪(2 mm深度)和肝臟(3 mm深度)組織,直接測量氧分壓(pO?)。校準基于21% O?飽和溶液和無氧溶液,每只動物重復3次取均值,確保數據精準。
技術優勢:
直接定量:克服免疫組化等間接方法的定性局限,提供連續pO?數值(mmHg),實時反映生理狀態下的缺氧程度。
高分辨率:25 μm超細探針定位特定組織區域(如肝臟左葉),減少解剖變異干擾,優于宏觀缺氧標記。
研究意義解讀:
揭示缺氧動態與病理時序:
數據(圖3)顯示性腺脂肪pO?在肥胖8周即顯著下降(對照組~40 mmHg → HFHFD 8周↓~18.9 mmHg),早于肝臟pO?下降(16周↓)。意義:證實脂肪缺氧是肥胖早期事件,驅動后續代謝紊亂;肝臟缺氧滯后但發生于MASLD早期,提示微循環障礙是脂肪變性向炎癥轉化的關鍵橋梁。
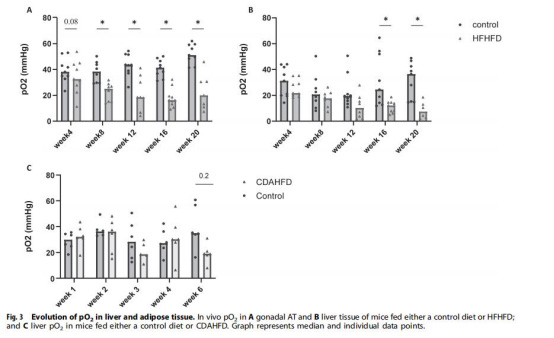
CDAHFD模型雖誘導嚴重MASH(NAS=8),但肝臟pO?無顯著變化(圖3C)。意義:證明肝臟缺氧非單純肝損傷所致,而依賴肥胖全身代謝紊亂(如脂肪缺氧→脂解↑→肝脂質沉積→血流障礙),確立肥胖在MASLD的核心地位。
驗證缺氧的分子機制:
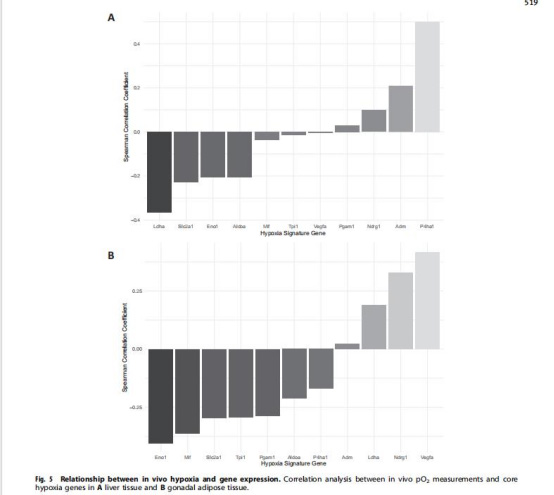
pO?↓與核心缺氧基因表達強相關(圖5):如脂肪pO?↓ ? Slc2a1(葡萄糖轉運體)↑,肝臟pO?↓ ? Ldha(乳酸脫氫酶)↑。意義:直接生理數據為基因表達提供驗證,揭示缺氧通過增強糖酵解(Slc2a1、Ldha)促進代謝重編程,解釋肥胖中能量代謝失調的機制。
支撐臨床轉化:
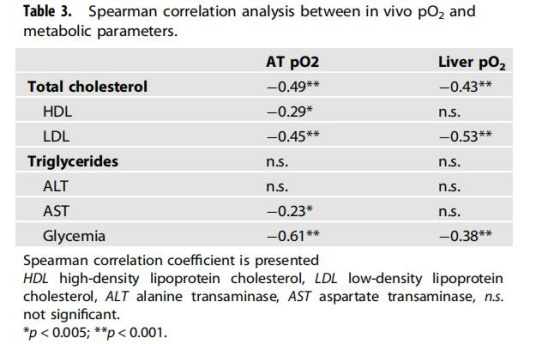
pO?與代謝參數顯著相關(表3):如脂肪pO?↓與血糖↑(r=-0.61)、LDL↑(r=-0.45)直接關聯。意義:確立缺氧作為代謝紊亂的早期生物標志物,為無創檢測(如血氧成像)提供依據;靶向缺氧(如HIF-1α抑制劑)或可阻斷肥胖向MASLD進展。
模型生理相關性:
pO?數據與組織學(pimonidazole染色)和基因表達一致,意義:驗證Unisense電極作為缺氧研究的金標準,推動其在臨床前研究的應用(如藥物療效評估)。
總體貢獻:Unisense微電極數據不僅量化了肥胖中缺氧的動態演變,更揭示了脂肪-肝臟軸的病理時序,為開發針對缺氧的精準干預策略奠定基礎。