熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
來源:Alzheimer’s Dement. 2024;20:890–903.
1. 論文摘要核心內容
核心發現:慢性高血壓通過血管緊張素II(Ang II)信號通路導致海馬小動脈(HA)功能障礙(內皮損傷、過度收縮),引發海馬低灌注(血流減少50%)和記憶損傷。ACE抑制劑卡托普利可逆轉上述病變,而抗氧化劑夾竹桃麻素(Apocynin)無效。
結論意義:海馬血管功能障礙是高血壓相關認知障礙(VCI)的關鍵機制,靶向Ang II信號可保護認知功能。
2. 研究目的
探究慢性高血壓伴隨衰老如何損害海馬血管功能及認知,并驗證以下假設:
高血壓隨年齡進展導致海馬血流減少、血管功能障礙和記憶衰退;
Ang II和氧化應激是核心機制,ACE抑制劑或抗氧化劑可干預。
3. 研究思路
年齡梯度對比:
分組:雄性自發性高血壓大鼠(SHR)分早期(4-5月)、中期(8-9月)、晚期(14-15月) vs. 正常血壓Wistar大鼠(4-5月)。
檢測:記憶行為學、海馬血流、離體海馬小動脈(HA)功能。
機制干預:
晚期SHR接受卡托普利(ACE抑制劑)或夾竹桃麻素(抗氧化劑)治療3個月。
檢測:治療后上述指標變化。
4. 測量數據及研究意義(標注圖表來源)
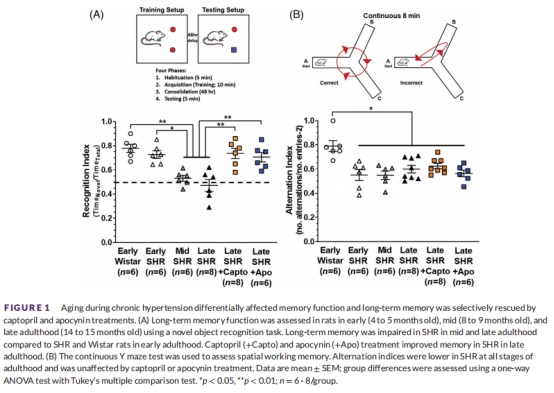
(1) 記憶功能(圖1)
數據:
長期記憶(NOR測試):SHR晚期識別指數顯著↓(圖1A)。
空間工作記憶(Y迷宮):所有SHR均受損(圖1B)。
意義:高血壓選擇性損害海馬依賴性長期記憶,且隨年齡加重。
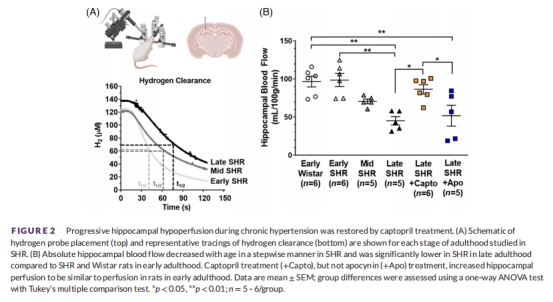
(2) 海馬灌注(圖2,表1)
數據:
晚期SHR海馬血流↓50%(vs. 早期SHR/Wistar)(圖2B)。
卡托普利恢復血流,夾竹桃麻素無效(圖2B)。
意義:海馬低灌注是記憶損傷的直接原因,且依賴Ang II信號。
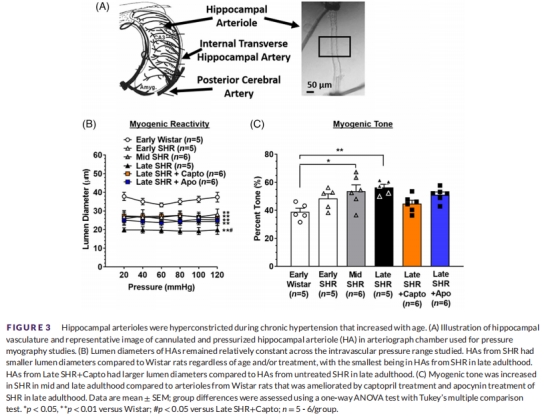
(3) 海馬小動脈(HA)功能
結構重塑(表S1):
晚期SHA管腔↓、壁厚↑(內向肥厚性重塑)。
肌源性緊張度(圖3C):
中期/晚期SHR肌源性緊張度↑(卡托普利和夾竹桃麻素均改善)。
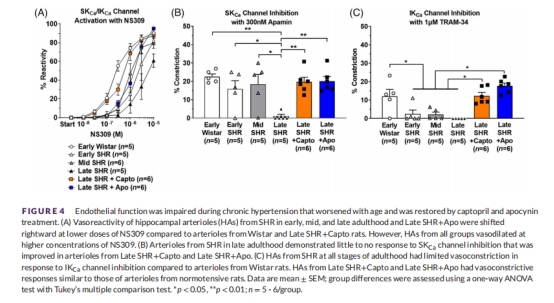
內皮功能(圖4):
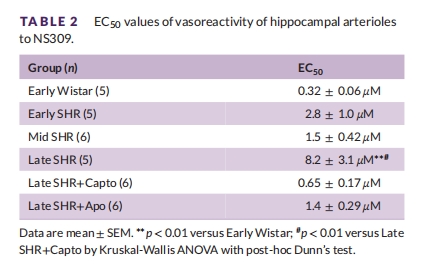
SKCa/IKCa通道功能(NS309反應):晚期SHR反應↓(EC50↑,表2),卡托普利完全恢復。
基底通道活性(Apamin/TRAM-34):晚期SHR無收縮反應(圖4B-C),治療組恢復。
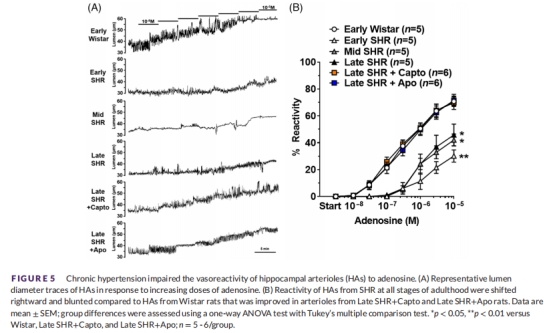
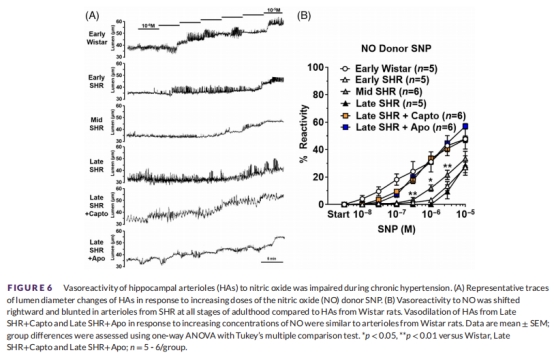
神經血管耦合(NVC)反應(圖5-6):
腺苷/NO介導的血管舒張:所有SHR均受損,治療組改善。
意義:Ang II導致內皮依賴性舒張功能障礙,損害NVC和血流調節。
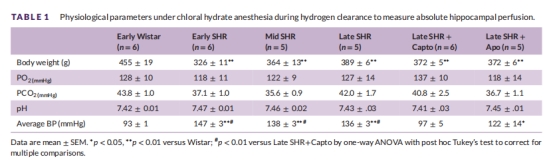
(4) 血壓與生理參數(表1)
卡托普利降壓,夾竹桃麻素無降壓作用。
5. 核心結論
機制鏈條:慢性高血壓 → Ang II信號激活 → 海馬小動脈內皮功能障礙/過度收縮 → 海馬低灌注 → 記憶損傷。
干預價值:ACE抑制劑通過抑制Ang II,改善血管功能、恢復血流、挽救記憶;抗氧化劑僅改善血管功能但無法恢復灌注或記憶。
臨床啟示:靶向海馬血管Ang II信號是防治高血壓相關認知障礙的有效策略。
6. 丹麥Unisense電極測量數據的詳細解讀
技術原理
方法:使用Unisense氫微電極(50μm尖端)植入海馬CA1區,通過氫清除法測量組織脫飽和半衰期(t1/2),計算絕對血流量(公式:血流 = (0.693 × 100g × 60s) / t?/?)。
優勢:高空間分辨率(定位CA1區)、實時動態監測、定量絕對血流值(mL/100g/min)。
關鍵數據(圖2, 表1)
發現:
晚期SHR海馬血流↓50%(圖2B),與HA過度收縮(圖3C)和記憶損傷(圖1A)同步。
卡托普利使血流恢復至早期水平,夾竹桃麻素無效(圖2B)。
生理參數保障:實驗過程中血氧(PO?)、二氧化碳(PCO?)、pH維持正常(表1),排除代謝干擾。
研究意義
首次在體定量高血壓衰老模型的海馬低灌注:
明確血流減少程度(50%)及時程(晚期最顯著),為"血管性認知障礙"提供直接生理證據。
建立血管-認知因果關聯:
低灌注與記憶損傷時空同步(圖1A vs. 圖2B),且血管功能改善(卡托普利組)伴隨血流與認知同步恢復。
揭示Ang II的核心作用:
卡托普利恢復血流而夾竹桃麻素無效,證明血流調節依賴Ang II而非單純氧化應激。
空間特異性價值:
CA1區對缺血高度敏感,此區域特異性低灌注直接關聯記憶損傷。
總結
本研究通過多尺度方法(行為學、在體血流、離體血管功能)闡明Ang II介導的海馬血管功能障礙是高血壓認知損傷的核心機制。丹麥Unisense電極提供的高精度海馬血流數據是關鍵證據,將分子機制(Ang II)與宏觀功能(認知)直接關聯,為ACE抑制劑的臨床應用提供了理論基礎。